SIFT4G的安装与使用
最近一个月状态有点不太对,有点控制不住自己的情绪。以后要是心情烦躁就写写博客吧!还能巩固知识。
SIFT4G的前身是sift,都是同一个东西,用来评估基因突变的有害程度,基于氨基酸序列的同源性和物理性质来预测氨基酸的替换对蛋白质功能是否影响。sift在网站上使用,它提供少量物种的在线查询,包括人类等。而SIFT4G则是本地版,当然windows和linux都有对应的工具可以运行,这里我们只讲述linux上SIFT4G的使用。
一、SIFT4G的安装
1.1本地编译安装
SIFT4G基于C++,软件编译需要4.9+版本的g++编译器,很不凑巧,我们服务器上只有4.8.5版本的g++编译器。
我们可以通过conda安装4.9+以上版本的g++,我们这里选择了5.4.0版本的编译器。如果你是直接通过conda在线安装,建议选一个新的环境,不然可能会有库文件依赖问题(下图所示)。我当时是通过conda本地安装在dcfpy3环境下,现在忘了细节了,不过google肯定能解决问题的。

在编译中发现一个问题,不知道安装还是什么其他原因,虽然dcfpy3这个子环境的g++是5.4.0版本。但是我直接编译还是会提示找不到库文件的问题,后来发现直接添加动态链接库可解决。
# 这些文件的地址需要按照自己的安装目录去更改,是不能直接套的
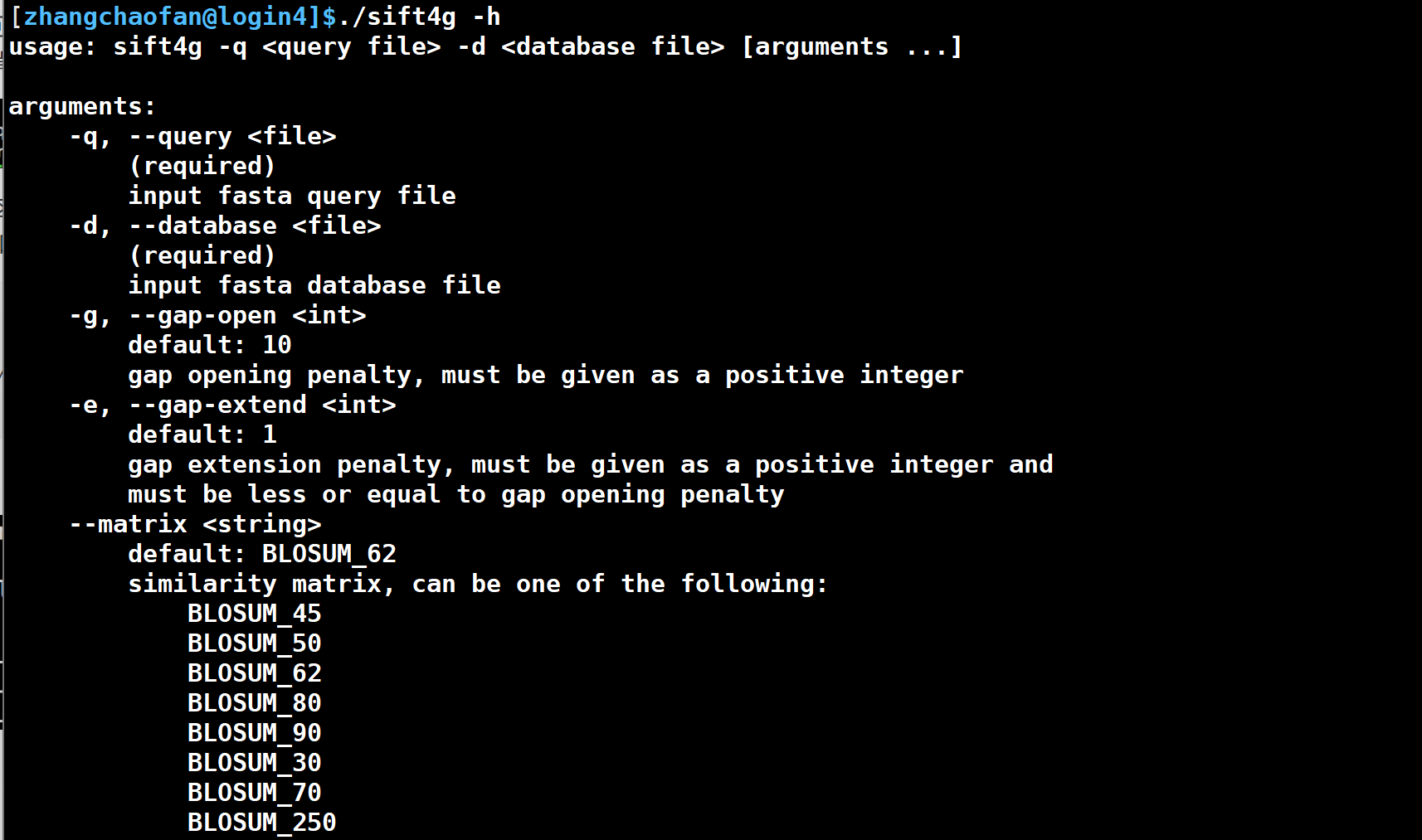
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/public/home/chaofan/anaconda3/envs/dcfpy3/lib出现下图这个就代表你安装好了,如果还报错可以去作者的github上问问,作者还是挺热情的。
1.2 conda安装
当然,你也可以直接通过conda在线安装,省时省事。(强烈建议新建一个环境来整)
conda install sift4g -c bioconda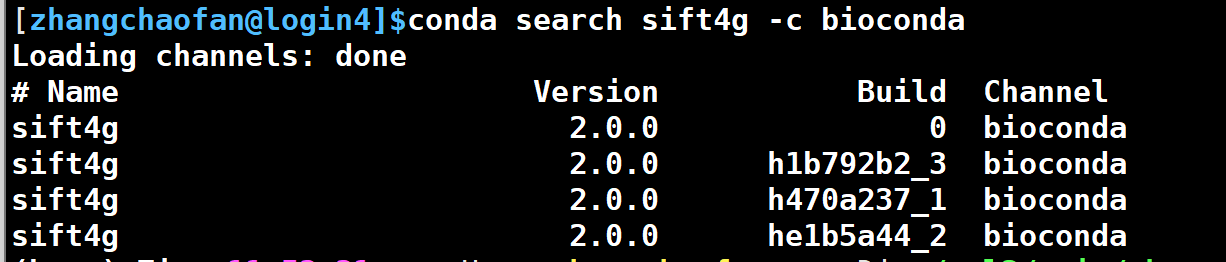
二、SIFT4G的使用
SIFT4G的使用非常简单,他的输入主要有两个,一个是针对特定物种的数据库,和该物种的蛋白组有关,另外一个则是VCF文件了,这里指的是SNP的,包含indel信息的应该也可以。特定物种的数据库可以去官网下载SIFT4G下载,然后直接使用SIFT4G Annotator进行注释就好了,没有的话就需要自己构建了。我选用的物种刚好没有,所以下面我们开始自己构建小麦的注释数据库。
2.1 SIFT4G注释数据库的生成
软件作者提供生成注释数据库的脚本,当然,你也可以直接联系软件作者帮你弄,需要将提供帮助构建数据库的人加入到你的论文作者中。注释脚本我们直接从Github上下载。主要是一个perl脚本,运行起来非常简单,直接用一行shell运行就行了。
生成这个注释数据库需要三个东西,包括注释物种的基因组序列fa和对应的gff文件,还需要一个非冗余的蛋白质数据库,你可以使用NCBI的nr蛋白质数据库或者Uniport的nr蛋白数据库。强烈建议构建这个注释数据库的时候拆分为不同的染色体来运行,可以并行化,速度提升非常的大。这个软件在构建过程中有一步需要根据gff和基因组的DNA序列来提取蛋白质,这一步非常非常的卡时间,有一个过程会一直往内存中加东西,但CPU的占用非常低,我之前整整个小麦的,结果运行了两天感觉没啥变化,后来分染色体体运行就非常快了。
非冗余数据库下载非常慢,这个文件非常大,下载过程中要注意下载不成功的情况。我们服务器上刚好有师兄弄好的,那我们直接用了。
构建注释数据库的命令非常简单,以染色体7D为例:
perl make-SIFT-db-all.pl -config ./wheat/chr7D.txt我们这里讲一下这个./wheat/chr7D.txt配置文件(一个#开头代表原来的注释):
## 氨基酸编码表格 这里使用标准的 当然也有线粒体什么的可以选
GENETIC_CODE_TABLE=1
GENETIC_CODE_TABLENAME=Standard
## 构建注释数据库的输入文件地址,里面需要包含特定的文件结构
PARENT_DIR=./wheat/Chr7D
ORG=wheat
## 最后输出的结果文件前缀 *.gz *.regions
ORG_VERSION=chr7D
#Running SIFT 4G
## sift4g软件的地址path,这里我们已经加入到PATH,不用加全局路径
SIFT4G_PATH=sift4g
## 非冗余蛋白库,比对的时候要用
PROTEIN_DB=/vol3/agis/TMP_group/chaofan/00.wenwen/02.database/ncbiNR/nr.fa
## 这里用默认的就好了,不用改
# Sub-directories, don't need to change
GENE_DOWNLOAD_DEST=gene-annotation-src
CHR_DOWNLOAD_DEST=chr-src
LOGFILE=Log.txt
ZLOGFILE=Log2.txt
FASTA_DIR=fasta
SUBST_DIR=subst
ALIGN_DIR=SIFT_alignments
SIFT_SCORE_DIR=SIFT_predictions
SINGLE_REC_BY_CHR_DIR=singleRecords
SINGLE_REC_WITH_SIFTSCORE_DIR=singleRecords_with_scores
## 这里用默认的就好了,不用改
# Doesn't need to change
FASTA_LOG=fasta.log
INVALID_LOG=invalid.log
PEPTIDE_LOG=peptide.log我们来讲一下PARENT_DIR=./wheat/Chr7D类似的文件结构:
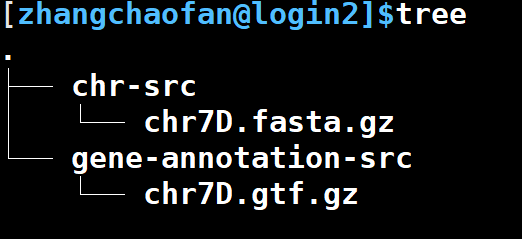
你需要在每一个“Chr7D”目录下放两个文件夹,名字应该和上图一样。chr-src放基因组序列,fasta格式,gene-annotation-src下面放对于的gz压缩的gtf文件,好像如果不是gz文件,后面有一步会报错,我也忘了具体的了,gz格式准没错。
这些都整好之后我们就可以用之前下载的注释脚本Github来构建我们的数据库了。
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/public/home/chaofan/anaconda3/envs/dcfpy3/lib
# 这个make-SIFT-db-all.pl 可以放到PATH里,也可以直接上全局路径
perl make-SIFT-db-all.pl -config ./wheat/chr7D.txt拆分为单个染色体后注释的时间很快的,我们成功结束的话目录下应该有下图这些东西。我们最后需要用的文件就在chr7D里面,chr7D.gz和chr7D.regions。
2.2 给vcf文件注释,获得有害变异
我们在构建完我们的注释数据库后,可以直接对vcf文件进行注释了。使用的软件为SIFT4G Annotator,这个软件有一个我非常理解不了的东西,为什么这个软件不能直接对gz文件进行操作?还得把原来压缩好的文件解压出来,再注释,再压缩。还有一个,这个软件会强制修改你vcf文件的染色体名称,它会去掉chr*前面的chr字符,比如你开始是chr7D,它后面操作的时候就变成了7D。这个会导致什么呢,这个的设计本来是为了多条染色体的,它会通过这个修改名称后的染色体名字去注释数据库找染色体对应的注释文件。所以上一步获得的chr7D.gz和chr7D.regions得修改成7D.gz和7D.regions才行哦。
这个问题我之前问过作者了,他当时也忘了,以为我vcf文件的染色体名称混入了其他的名称。后来我翻他源码才找到了,头疼。第174到177步就是删除染色体名称中“chr”(得是chr开头的哦)的操作代码。

在整完前面这些步骤后,我们开始注释了:
java -jar /vol3/agis/TMP_group/chaofan/09.SIFT/sift4g/bin/SIFT4G_Annotator.jar -c -i chr7D.vcf -d /vol3/agis/TMP_group/chaofan/09.SIFT/scripts_to_build_SIFT_db/wheat/Chr7D/chr7D -r . -t我们已经整完了所有的步骤了,-i 待注释的vcf文件, -c 命令行运行模式, -d 之前生成的注释数据库文件夹地址,-r 输出文件地址 -t 提取多个转录本的注释(可选)。下一次更新jcvi的使用,这几天刚好在学习这个,做共线性分析的。


