这是一个交链孢霉属物种二代数据基因组组装注释流程
0.环境准备
conda create -n genome_assemble
conda activate genome_assemble
conda install fastqc fastuniq bwa kmergenie jellyfish quast -y -c bioconda
# 进入到自己的软件文件夹,没有就新建一个: mkdir software
cd software
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/3.0.2/sratoolkit.2.11.1-centos_linux64.tar.gz
tar -xvzf sratoolkit.2.11.1-centos_linux64.tar.gz
wget http://cab.spbu.ru/files/release3.15.5/SPAdes-3.15.5-Linux.tar.gz
tar -xvzf SPAdes-3.15.5-Linux.tar.gz
wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.39.zip
unzip Trimmomatic-0.39.zip
# 下载interproscan
wget https://ftp.ebi.ac.uk/pub/software/unix/iprscan/5/5.61-93.0/interproscan-5.61-93.0-64-bit.tar.gz
tar -zxvf interproscan-5.61-93.0-64-bit.tar.gz
# 加入环境变量
export PATH="/data/chaofan/software/sratoolkit.2.11.1-centos_linux64/bin:$PATH"
export PATH="/data/chaofan/software/SPAdes-3.15.5-Linux/bin:$PATH"
export PATH="/data/chaofan/software/interproscan-5.61-93.0:$PATH"
1.下载数据
# 设置你自己的工作路径,把数据什么的都放在单独的一个工作路径
workdir=/data/pipelines/01.Gene_anno/01.genome_assemble/01.Alternaria_sp
cd $workdir
mkdir 00.raw_data && cd 00.raw_data
nohup prefetch SRR16888499 -O ./ & # RNA-seq
nohup prefetch SRR16914355 -O ./ & # WGS(Whole Genome Sequencing)2.质控
2.1 sra2fastq
fastq-dump --split-3 SRR16888499/SRR16888499.sra &
fastq-dump --split-3 SRR16914355/SRR16914355.sra &
2.2 raw_fastqc
fastqc SRR16888499_1.fastq SRR16888499_2.fastq SRR16914355_1.fastq SRR16914355_2.fastq -t 4 -o ./这个是SRR16888499(RNA-seq)的测序质量值和接头报告,我们发现这个质量是非常不错的,但还是使用Trimmomatic对raw_reads进行过滤。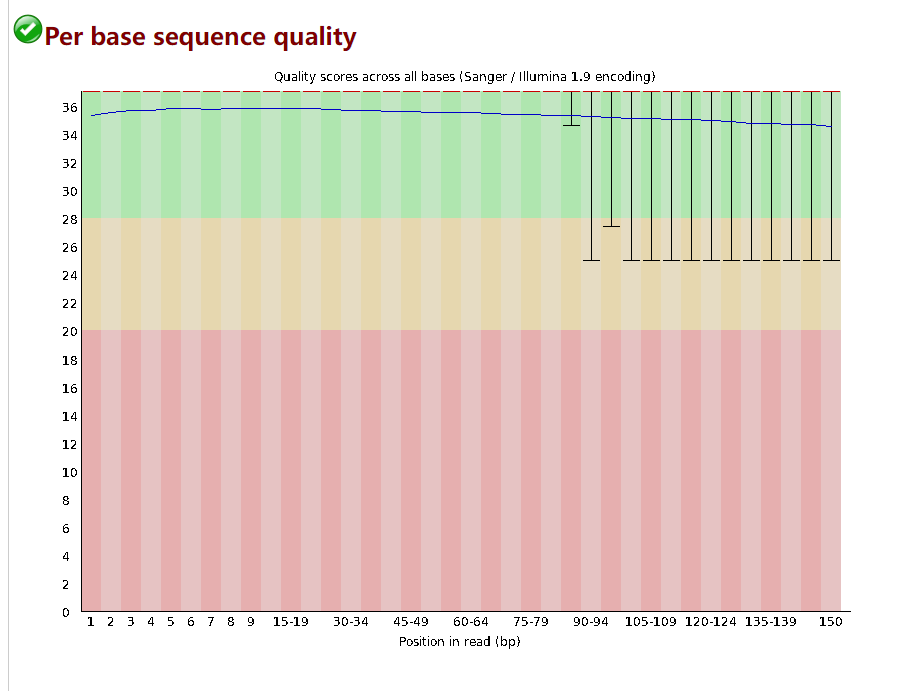
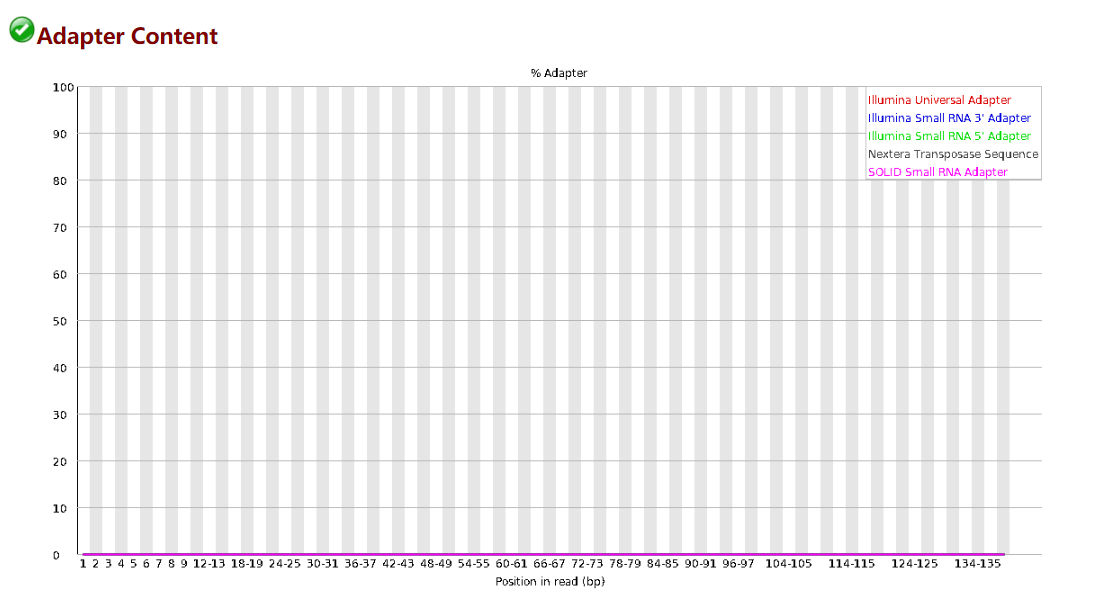
SRR16914355的质量也很好,我们按同一标准过滤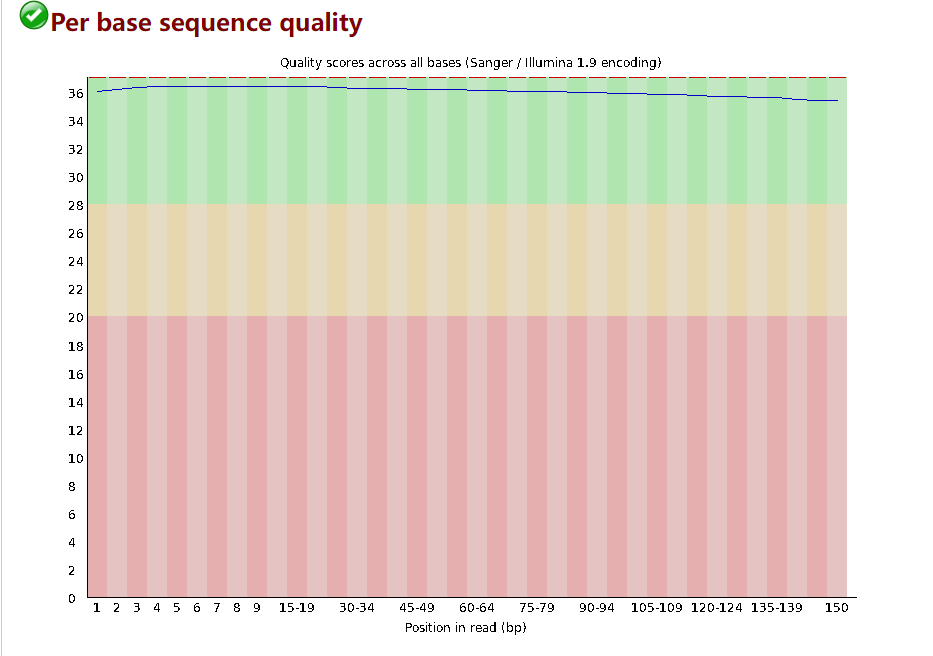
2.3 Trimmomatic filter
注意命令和文件的地址,要换回自己的地址,别傻乎乎的直接用
# 将不同的测序引物序列合并为一个fasta文件,这样可以处理不同的测序文件
cat /data/chaofan/software/Trimmomatic-0.39/adapters/*.fa > /data/chaofan/software/Trimmomatic-0.39/adapters/all.fa
# cat完之后检查一下 all.fa 是否是标准的fasta格式,有几个adapter文件末端好像没有换行符,导致cat后 fasta的> + ID的那行接到了上一条序列的序列行了
#
java -jar /data/chaofan/software/Trimmomatic-0.39/trimmomatic-0.39.jar PE -phred33 SRR16888499_1.fastq SRR16888499_2.fastq SRR16888499_1.paired.fastq SRR16888499_1.unpaired.fastq SRR16888499_2.paired.fastq SRR16888499_2.unpaired.fastq ILLUMINACLIP:/data/pipelines/00.RNA_seq/Trimmomatic-0.39/adapters/all.fa:2:30:10:1:TRUE SLIDINGWINDOW:4:20 LEADING:3 TRAILING:3 MINLEN:40 -threads 30
# SRR10283202_1.paired.fastq SRR10283202_2.paired.fastq就是我们TRANSCRIPTOMIC的clean data
# 这里改下名字,不然后面fungap会找不到(只找固定后缀文件)
mv SRR16888499_1.paired.fastq SRR16888499_1.fq
mv SRR16888499_2.paired.fastq SRR16888499_2.fq
java -jar /data/chaofan/software/Trimmomatic-0.39/trimmomatic-0.39.jar PE -phred33 SRR16914355_1.fastq SRR16914355_2.fastq SRR16914355_1.paired.fastq SRR16914355_1.unpaired.fastq SRR16914355_2.paired.fastq SRR16914355_2.unpaired.fastq ILLUMINACLIP:/data/pipelines/00.RNA_seq/Trimmomatic-0.39/adapters/all.fa:2:30:10:1:TRUE SLIDINGWINDOW:4:20 LEADING:3 TRAILING:3 MINLEN:40 -threads 30
# SRR22980490_1.paired.fastq SRR22980490_2.paired.fastq就是我们WGS的clean data,但还要去一下重复序列
# 参数的具体含义建议去官网上搜索,这里不详细讲述了,参数选择并不唯一2.4 remove PCR duplication
在基因组组装前,需要对WGS的测序数据去除PCR重复,这里使用fastuniq操作。虽然raw_data就已经没什么重复序列了,但我们还是去一遍,转录组数据一般不用去除这些重复序列。
mkdir -p ../01.clean_data && mv *.paired.* ../01.clean_data
cd ../01.clean_data
# Remove exact duplicates
ls SRR16914355_1.paired.fastq SRR16914355_2.paired.fastq > sample_lst
fastuniq -i sample_lst -o SRR16914355_1.rd.fastq -p SRR16914355_2.rd.fastq 2.5 clean_fastqc
fastqc SRR16914355_1.rd.fastq SRR16914355_2.rd.fastq SRR16888499_1.fq SRR16888499_2.fq -t 4 -o./这里就不放图了,不知道上传的是不是就是clean_data,质量很好。
3.基因组组装
3.1 genome survey
kmer在生物信息学上有非常广泛的运用,有非常多方法都是基于kmer的,感兴趣的自己去了解下吧。这里主要使用kmergenie来分析。
ls SRR16914355_1.rd.fastq SRR16914355_2.rd.fastq > kmer_lst
kmergenie kmer_lst -o kmergenie_result/first_kmer -k 127 -l 27 -s 6 -t 40kmergenie log
running histogram estimation
File sample_lst starts with character "S", hence is interpreted as a list of file names
Reading 2 read files
Linear estimation: ~1910 M distinct 74-mers are in the reads
K-mer sampling: 1/364
| processing |
[going to estimate histograms for values of k: 127 121 115 109 103 97 91 85 79 73 67 61 55 49 43 37 31
------------------------------------------------------------------
----------------------------------------------------Total time Wallclock 1924.2 s
fitting model to histograms to estimate best k
estimation of the best k so far: 109
refining estimation around [103; 115], with a step of 2
running histogram estimation
File sample_lst starts with character "S", hence is interpreted as a list of file names
Reading 2 read files
Linear estimation: ~2066 M distinct 68-mers are in the reads
K-mer sampling: 1/394
| processing |
[going to estimate histograms for values of k: 115 113 111 109 107 105 103
----------------------------------------------------------------------------------------------------------------------Total time Wallclock 798.288 s
fitting model to histograms to estimate best k
table of predicted num. of genomic k-mers: kmergenie_result.dat
recommended coverage cut-off for best k: 1
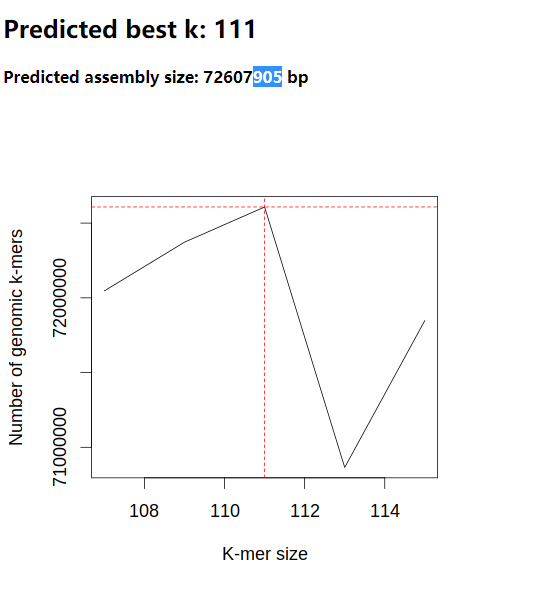
best k: 111最佳K=111,我们再跑一遍kmergenie,这次缩小下范围
kmergenie kmer_lst -o kmergenie_result/re_kmer -k 115 -l 107 -s 2 -t 40这次最佳还是K=111,后续我们使用SPAdes组装也要添加这个k值。预估的基因组大约72,607,905bp。
这里是NCBI上公布的基因组序列信息,大小和预估出来的差不太多。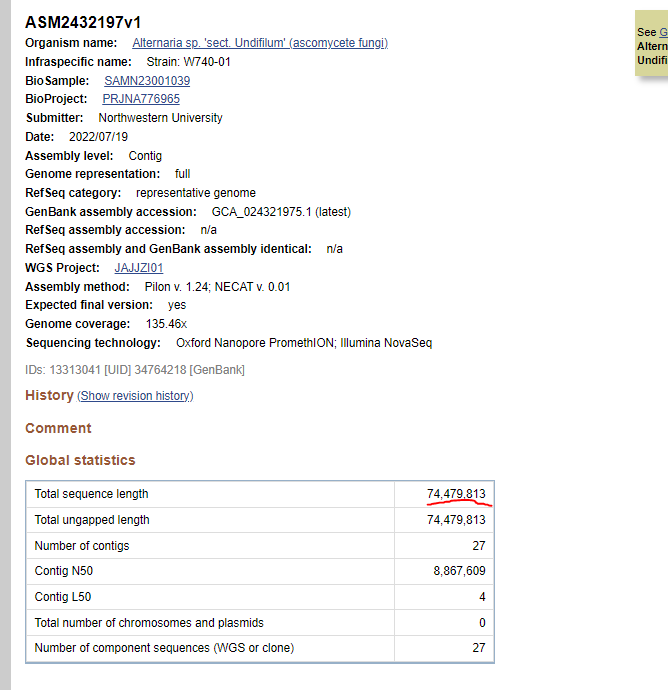
3.2 SPAdes assemble
主要使用spades来进行基因组组装。软件的具体参数还是建议去GitHub上自己看,直接用这个也行。spades要求kmer长度必须为奇数。
nohup /data/chaofan/software/SPAdes-3.15.5-Linux/bin/spades.py -k 21,33,55,77,99,109,111,127 -t 20 --careful --cov-cutoff auto -1 SRR16914355_1.rd.fastq -2 SRR16914355_2.rd.fastq -o ../02.draft_genome &3.3 quast评估基因组组装质量
cd ../02.draft_genome
conda activate genome_assemble
/data/chaofan/miniconda/envs/genome_assemble/bin/quast scaffolds.fasta
conda deactivate这个是单纯的二代测序数据的组装结果,N50 126.245kbp。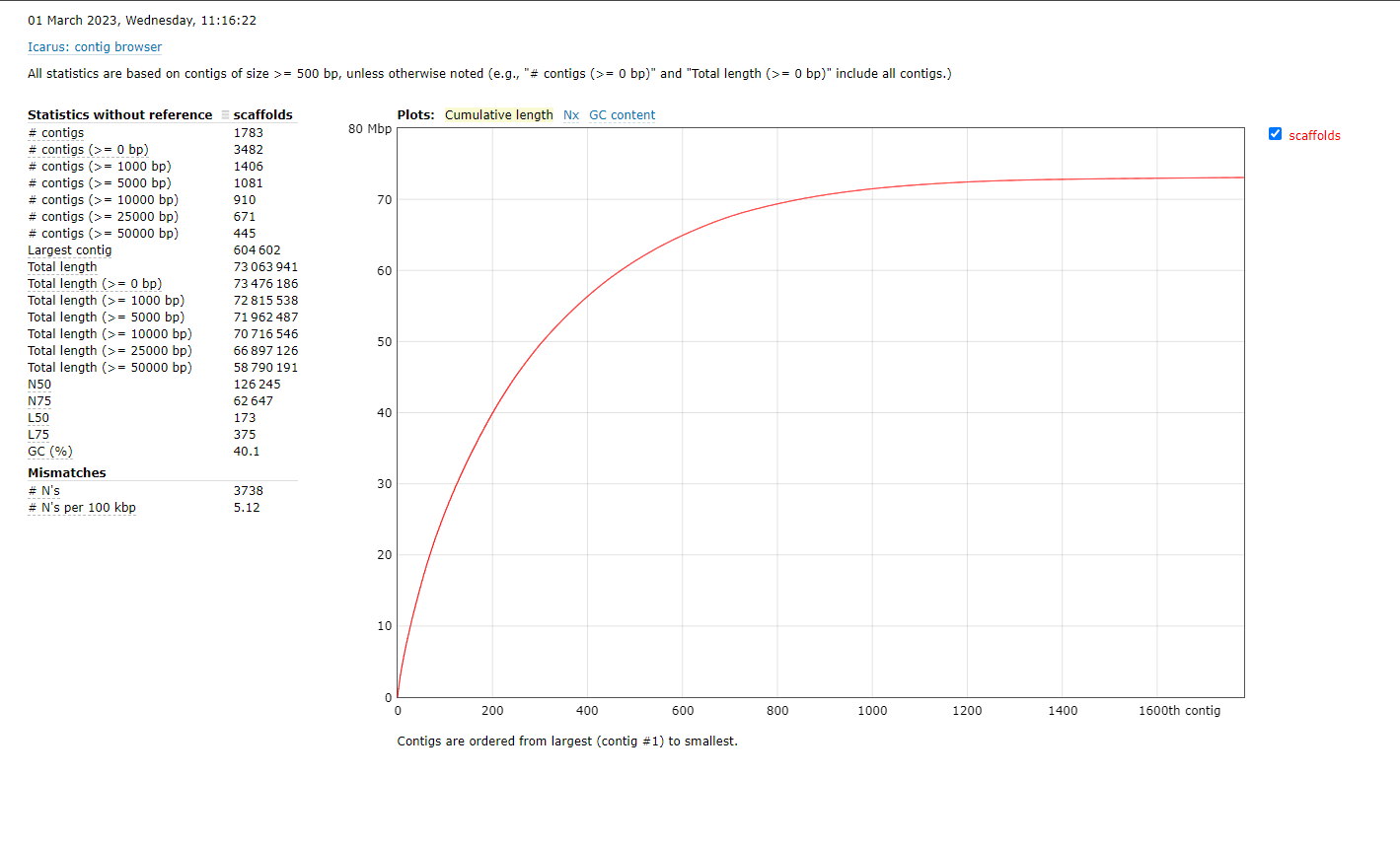
3.4 BUSCO评估组装一致性
# 单独新建一个环境配置
# busco 5.4.4有一个BUG https://gitlab.com/ezlab/busco/-/issues/627
# 很遗憾 busco 5.4.5运行这个DB:dothideomycetes_odb10也会报错(emmm 只有在对基因组使用这个DB评估的时候才有问题,注释蛋白的评估是没问题的) 请安装较老一点版本的busco(5.2.*)。 这里我们直接换一个DB吧
conda create -n busco.5.4.5 -c conda-forge -c bioconda busco=5.4.5
conda activate busco.5.4.5
busco -c 8 -i scaffolds.fasta -l /data/chaofan/source/busco_db/fungi_odb10 -m geno -o busco.5.4.5_fungi_odb10
conda deactivate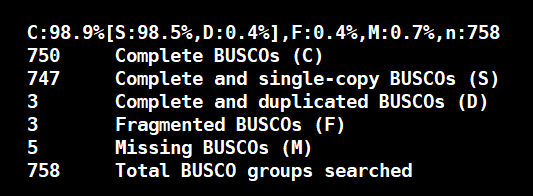
4.基因注释
基因注释主要使用FunGAP来做,如果不通过docker安装的话,在使用过程中会遇到各种各样的错误,善用google|bing,一般都是可以解决的(这应该是整个过程中最折腾的)。我这里新建了一个conda环境用来基因注释,所以这里我们先切换下环境:
conda activate fungapFunGAP的安装请遵循Github,这里就不展开讲了
基因注释主要需要三个输入:
- 组装好的基因组文件;
- 近源物种的蛋白序列;
- August预测 物种模型的选择;
- 最好测个转录组, FunGAP强制必须加上RNA-seq数据,无论是clean_data还是比对后的bam文件(这个可以修改,但是大部分的脚本文件都需要相应的调整,也可以从NCBI上下载RNA-seq数据)。
4.1 获取近源物种的蛋白
FunGAP提供了脚本,可以直接从ncbi下载近源物种的蛋白序列。你可以在你的FunGAP文件夹下面找到download_sister_orgs.py,这里我们通过全局路径调用。如果可以的话,尽量下载同属或者同科的所有蛋白序列。
cd $workdir
mkdir 03.gene_anno && cd 03.gene_anno
python /data/pipelines/01.Gene_anno/00.FunGAP/FunGAP/download_sister_orgs.py \
--download_dir sister_orgs \
--taxon "Alternaria" \
--num_sisters 4 \
--email_address 2439555626@qq.com
zcat sister_orgs/*faa.gz > prot_db.faa- –taxon 物种名或属名 它会通过ncbi的Taxonomy遍历近源物种的Assembly_data去下载已公布基因组的蛋白数据;
- –num_sisters 下载的近源物种的数量,一般来说越多越好;
- –email_address 邮箱地址,随便填.
4.2 挑选August预测的物种模型
python /data/pipelines/01.Gene_anno/00.FunGAP/FunGAP/get_augustus_species.py \
--genus_name "Alternaria" \
--email_address 2439555626@qq.com
# --genus_name 物种名或属名 依然通过ncbi的Taxonomy找到
# 已有的近源物种的augustus模型,可以直接用Suggestions:
We found them in subphylum level: Pezizomycotina
aspergillus_fumigatus
aspergillus_nidulans
aspergillus_oryzae
aspergillus_terreus
botrytis_cinerea
chaetomium_globosum
coccidioides_immitis
fusarium_graminearum
histoplasma_capsulatum
magnaporthe_grisea
neurospora_crassa这里我就简单的选了第一个直接用了。
4.3 FunGAP进行基因注释
emmmmmm,有一个隐藏的问题,maker train的过程中会用到TRF对序列进行处理,TRF应该设置了一个256个字符长度的数组来储存输入文件的全局路径,如果你的输入文件路径大于256,TRF就会buffer overflow detected,老倒霉蛋了。这个问题好像还是第一次发现的,特别是通过mpirun并行maker,会给输入文件加上一堆特殊含义的后缀什么的(这也和我改了maker的默认TMPDIR有关,我把它设置为当前运行路径下,可能当前全局路径本身就很长了)。
/data/pipelines/01.Gene_anno/00.FunGAP/FunGAP/fungap.py \
--genome_assembly ../02.draft_genome/scaffolds.fasta \
--trans_read_1 SRR22973808_1.fastq \
--trans_read_2 SRR22973808_2.fastq \
--augustus_species aspergillus_fumigatus \
--busco_dataset /data/chaofan/source/busco_db/dothideomycetes_odb10 \
--sister_proteome prot_db.faa \
--num_cores 32 \
--output_dir Alternaria4.4 BUSCO评估基因注释结果(这里用的dothideomycetes_odb10)
# 检查使用二代数据组装基因组基因注释的busco值
busco -c 8 -i Alternaria/fungap_out/fungap_out_prot.faa -l /data/chaofan/source/busco_db/dothideomycetes_odb10 -m prot -o Alternaria_dothideomycetes_odb10要是植物有这么高就好了。增加prot_db.faa里面同源物种的蛋白数量会适当的提高busco值,可能会加个百分之零点几吧。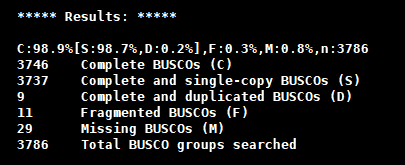
4.5 检查gff文件
前段时间被gff文件中的CDS phase搞了一波,以后如果我做完基因注释,绝对检查一遍。使用GFF3toolkit
gff3_QC -g ../03.gene_anno/Alternaria/fungap_out/fungap_out.gff3 -f scaffolds.fasta -o error.txt -s statistic.txt我们这里可能有一个小问题:
['Line 3'] Esf0003 Error [Strand information missing: legal chacracter, ".", found at the strand field]这个是由于我们的gff文件中还包含了序列长度信息行,这个可以不用管。假设你的gff确实有别的问题(注意看error.txt),则用gff3_fix进行矫正,gff3_fix也是属于GFF3toolkit的一个工具
gff3_fix -qc_r error.txt -g ../03.gene_anno/Alternaria/fungap_out/fungap_out.gff3 -og ../03.gene_anno/Alternaria/fungap_out/corrected.gff34.6 对注释蛋白进行功能注释
这里我们使用interproscan.sh来对注释的蛋白进行功能注释,并添加到gff文件中。
# 删除序列末尾的 * 号
sed -i 's/\*$//g' fungap_out_prot.faa
/data/chaofan/software/interproscan-5.61-93.0/interproscan.sh -i fungap_out_prot.faa -f tsv -appl Pfam --goterms -pa --iprlookup -b Alternaria --tempdir /data/pipelines/01.Gene_anno/maker_tmp/
# 将interproscan注释加到gff文件内
/data/pipelines/01.Gene_anno/00.FunGAP/FunGAP/gff3_add_pfam.py --input_gff3 fungap_out.gff3 --pfam_file Alternaria.tsv
# 默认注释是加在mRNA 第一个CDS行的第9列(key 列),你也可以直接修改这个脚本,把他加在mRNA的后面。
如有任何问题,恳请联系我修正。

