流程总览:

本流程参考华中农业大学李国亮老师的课程PPT,旨在提供一个打包好的pipeline,一行命令进行转录组数据的分析。
0. 环境配置
需要预安装的一些软件,包括:
condaconda create -n hzau_RNA-seq conda activate hzau_RNA-seq conda install python=3.10 fastqc fastp multiqc=1.26 sra-tools=3.1 samtools=1.21 -c bioconda -c conda-forge conda install hisat2=2.2.1 STAR=2.7.11b stringtie=2.2.3 subread=2.1.1 deeptools snakemake -c bioconda -c conda-forge # 大爷的 之前openssl版本有问题 conda install r-base=4.4.2 r-biocmanager r-ggplot2=3.5.1 curl=8.11.1 openssl=3.4.0 -c bioconda -c conda-forge # 我这里通过conda安装bioconductor-deseq2老有问题,只能通过BiocManager。可能你直接conda装就成功了 # conda install bioconductor-deseq2 -c bioconda -c conda-forgeinstall_DESeq2.R# 如DEseq2直接通过conda安装成功,则不需要运行 "BiocManager::install("DESeq2")" BiocManager::install("DESeq2") # 安装其它依赖 install.packages("purrr") install.packages("tibble") library(DESeq2) # Done!1. 原始数据下载
# 配置工作目录 mkdir 00.raw_sra 01.raw_fastq 02.clean_fastq 03.alignment_file 04.count_matrix 05.DEseq2 # 下载原始sra文件 cat SRR_Acc_List.txt | awk '{print $1}' |while read line; do prefetch ${line} -O 00.raw_sra & done ## 下载完需检查文件大小是否与原网页一致 # sra转fastq.gz cat SRR_Acc_List.txt | awk '{print $1}' | while read line; do fastq-dump 00.raw_sra/${line}/${line}.sra --split-3 --gzip -O 01.raw_fastq & doneSRR_Acc_List.txtSRR15859987 # mature leaves Col-0 Repeat1 SRR15859988 # mature leaves Col-0 Repeat2 SRR15859989 # mature leaves Col-0 Repeat3 SRR15859993 # mature leaves met1-3 Repeat1 SRR15859994 # mature leaves met1-3 Repeat2 SRR15859995 # mature leaves met1-3 Repeat32.
李老师这里介绍了几种常用的流程:Shell命令行操作- Fastp -> HISAT2/STAR -> featureCounts|htseq-count(基于gene Counts) -> DESeq2
- Fastp -> HISAT2/STAR -> StringTie(基于转录本) -> EdgeR
我这个()注释位置与原PPT不同,放这里个人感觉更直观。
2.1 数据质控
# raw_data 测序数据质量检测
fastqc 01.raw_fastq/*.gz -o 01.raw_fastq/ -t 12
multiqc 01.raw_fastq/ -o 01.raw_fastq/multiqc # 可以看下multiqc_report.html,原始数据有点接头
# fastq 测序数据质控
for i in 87 88 89 93 94 95
do
fastp -i 01.raw_fastq/SRR158599${i}_1.fastq.gz -o 02.clean_fastq/SRR158599${i}_1.fastq.gz -I 01.raw_fastq/SRR158599${i}_2.fastq.gz -O 02.clean_fastq/SRR158599${i}_2.fastq.gz -h 02.clean_fastq/SRR158599${i}.fastp.html -j 02.clean_fastq/SRR158599${i}.fastp.json --thread 16
done
# clean_data 测序数据质量检测
fastqc 02.clean_fastq/*.gz -o 02.clean_fastq/ -t 12
multiqc 02.clean_fastq/ -o 02.clean_fastq/multiqc # 对比下质控前后结果
# 目前大部分情况下是不需要去除rRNA的2.2 数据比对
首先下载基因组数据
# dna_sm 基因组softmask版本
wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-61/fasta/arabidopsis_thaliana/dna/Arabidopsis_thaliana.TAIR10.dna_sm.toplevel.fa.gz
wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-61/gtf/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.61.gtf.gz
gzip -d *.gz
mkdir Reference
mv Arabidopsis_thaliana.TAIR10.61.gtf Reference/
mv Arabidopsis_thaliana.TAIR10.dna_sm.toplevel.fa Reference/2.2.1 HISAT2比对
mkdir Reference/hisat2_index
# 构建HISAT2 基因组比对索引
hisat2-build -p 20 Reference/Arabidopsis_thaliana.TAIR10.dna_sm.toplevel.fa Reference/hisat2_index/Arabidopsis_thaliana.TAIR10
mkdir -p 03.alignment_file/hisat2_result
for i in 87 88 89 93 94 95
do
hisat2 -x Reference/hisat2_index/Arabidopsis_thaliana.TAIR10 -1 02.clean_fastq/SRR158599${i}_1.fastq.gz -2 02.clean_fastq/SRR158599${i}_2.fastq.gz -S 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.sam -p 20 --summary-file 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.summary
done 2.2.2 STAR比对
其实使用STAR比对这一步可以不做的,我个人一般也是直接使用HISAT2进行mapping。
mkdir Reference/STAR_index
# 构建STAR 基因组比对索引
STAR --runThreadN 16 --runMode genomeGenerate --genomeDir Reference/STAR_index/ --genomeFastaFiles Reference/Arabidopsis_thaliana.TAIR10.dna_sm.toplevel.fa
# 有个 WARNING啊,课件里只用了一条染色体应该也有WARNING的
# !!!!! WARNING: --genomeSAindexNbases 14 is too large for the genome size=119667750, which may cause seg-fault at the mapping step. Re-run genome generation with recommended --genomeSAindexNbases 12
# 我们重新生成
STAR --runThreadN 16 --runMode genomeGenerate --genomeDir Reference/STAR_index/ --genomeFastaFiles Reference/Arabidopsis_thaliana.TAIR10.dna_sm.toplevel.fa --genomeSAindexNbases 12
mkdir -p 03.alignment_file/star_result
for i in 87 88 89 93 94 95
do
STAR --runThreadN 16 --runMode alignReads --readFilesCommand zcat --outSAMunmapped None --genomeDir Reference/STAR_index/ --readFilesIn 02.clean_fastq/SRR158599${i}_1.fastq.gz 02.clean_fastq/SRR158599${i}_2.fastq.gz --outFileNamePrefix 03.alignment_file/star_result/SRR158599${i}_
done 2.2.3 sam2bam
HISAT2的输出sam是按照readsname进行排序的,可以直接用于htseq-count进行计数。但有些软件(比如:StringTie1)的输入要求必须是按position进行排序,我们需要使用samtools进行转换。而且sam文件非常大,将sam转换为bam也能很好的缓解储存压力。samtools sort的输出默认是按照position进行排序,如果这样的bam用于htseq-count计数,需指定-r pos(default: name)。这里只是提一嘴,我们这里不使用htseq-count进行计数。
for i in 87 88 89 93 94 95
do
samtools view -@ 16 -bS 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.sam > 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.bam && rm 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.sam
samtools sort -@ 16 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.bam -o 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.sort.bam && rm 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.bam
samtools index 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.sort.bam
done2.3 计算基因表达量
2.3.1 featureCounts|htseq-count
这里使用featureCounts或htseq-count都行,我们这里参照原PPT使用featureCounts。
mkdir -p 04.count_matrix/featureCounts
for i in 87 88 89 93 94 95
do
featureCounts -a Reference/Arabidopsis_thaliana.TAIR10.61.gtf -o 04.count_matrix/featureCounts/SRR158599${i}.featurecount.txt -p 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.sort.bam # 双端数据千万别忘了加 -p
done featureCounts的输出很简单,单个样本的情况下输出七列,第一列为基因ID,第六列为这个基因所有exon的长度(去掉了不同转录本间的冗余长度,可直接用来算FPKM),第7列为mapping到的Fragments数量(单端测序数据也称为reads数量)。

2.3.2 Stringtie
Stringtie可以用来鉴定新的转录本,挖掘可变剪切事件,也可以直接定量,参考简书教程2。
mkdir -p 04.count_matrix/stringtie
for i in 87 88 89 93 94 95
do
stringtie -p 16 03.alignment_file/hisat2_result/SRR158599${i}.hisat2.sort.bam -G Reference/Arabidopsis_thaliana.TAIR10.61.gtf -o 04.count_matrix/stringtie/SRR158599${i}.gtf -e -A 04.count_matrix/stringtie/SRR158599${i}.gene.tab
done注意,Stringtie的输出直接为FPKM和TPM,没有count。后续的DESeq2差异表达分析的输入必须使用原始的count。其实我们用了featureCounts|htseq-count进行计数后没必要再用Stringtie,如果需要FPKM和TPM值,可以通过count进行转换。

2.4 检查样本的重复性
# 计算每个bam文件的覆盖度情况
multiBamSummary bins --bamfiles 03.alignment_file/hisat2_result/SRR15859987.hisat2.sort.bam 03.alignment_file/hisat2_result/SRR15859988.hisat2.sort.bam 03.alignment_file/hisat2_result/SRR15859989.hisat2.sort.bam 03.alignment_file/hisat2_result/SRR15859993.hisat2.sort.bam 03.alignment_file/hisat2_result/SRR15859994.hisat2.sort.bam 03.alignment_file/hisat2_result/SRR15859995.hisat2.sort.bam --minMappingQuality 30 --labels Col_rep1 Col_rep2 Col_rep3 met_rep1 met_rep2 met_rep3 -out 03.alignment_file/readCounts.npz --outRawCounts 03.alignment_file/readCounts.tab -p 20
# 基于覆盖度信息进行层次聚类
plotCorrelation -in 03.alignment_file/readCounts.npz --corMethod spearman --skipZeros --plotTitle "Spearman Correlation of Read Counts" --whatToPlot heatmap --colorMap RdYlBu --plotNumbers -o 03.alignment_file/heatmap_SpearmanCorr_readCounts.png --outFileCorMatrix 03.alignment_file/readCounts.tab样本重复性不错: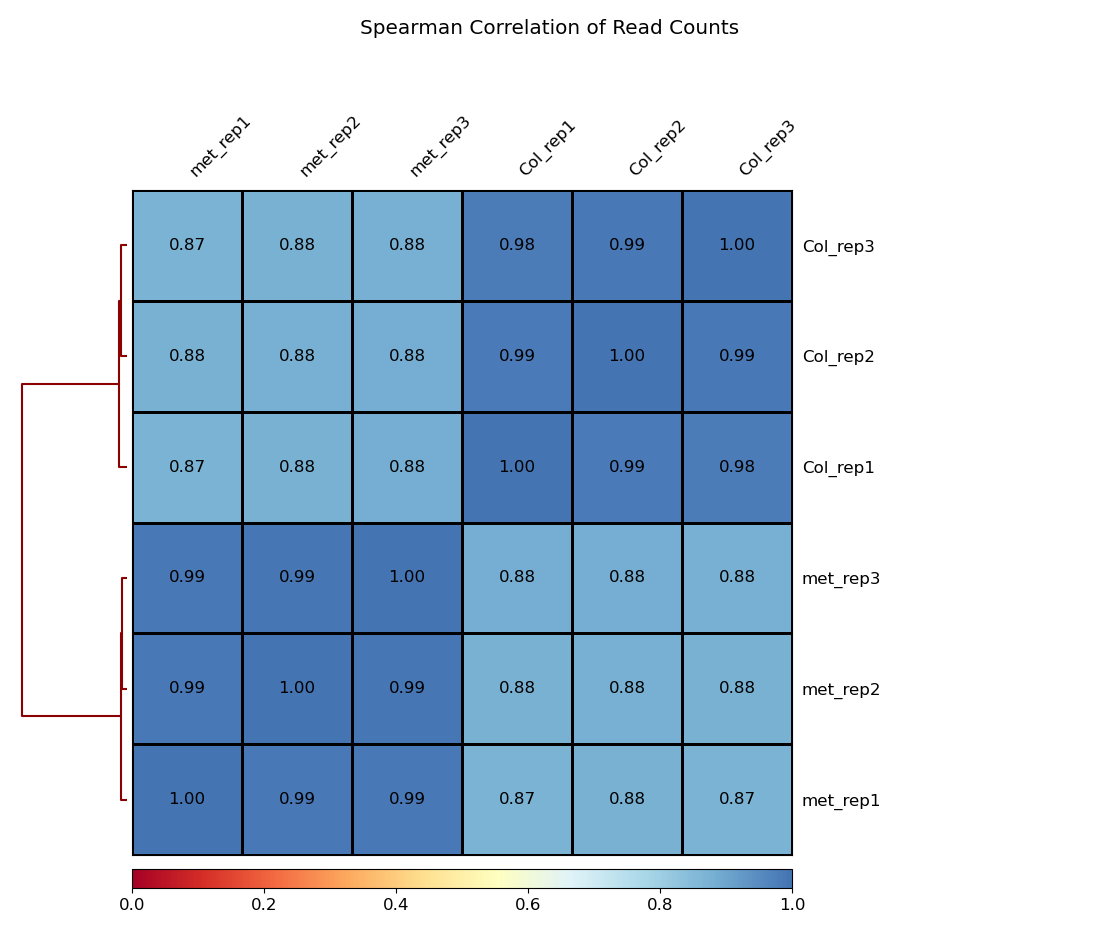
2.5 DESeq2差异表达分析
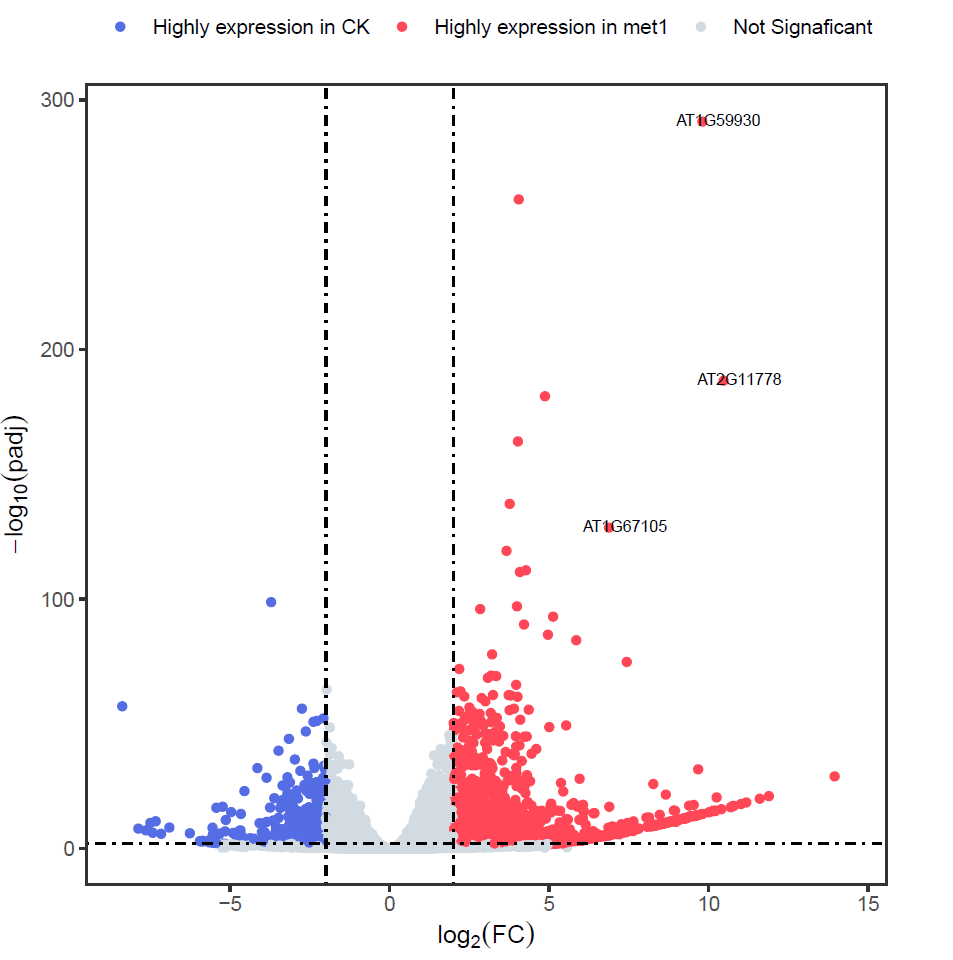
Rscript DEseq2.RDEseq2.Rlibrary(purrr) suppressMessages(library(DESeq2)) library(ggplot2) library(tibble) ################# Merge count table ################## samplelist <- c("SRR15859987", "SRR15859988", "SRR15859989", "SRR15859993", "SRR15859994", "SRR15859995") # 读取不同样本的基因表达量列表 exprlist <- lapply(samplelist, function(x){ expr <- read.table(paste0("04.count_matrix/featureCounts/", x, ".featurecount.txt"), header=T) ## 读取文件 expr<- expr[,c(1,7)] ### 提取基因id和表达量值 colnames(expr) <- c("GeneID", x) ## 重命名 expr }) # 多个基因样本的基因表达量列表合并 expr.mat <- purrr::reduce(exprlist, function(x,y){merge(x,y, by="GeneID", all.x=T)}) write.table(expr.mat, file = "./05.DEseq2/merged_count_matrix.txt", sep = "\t", quote = FALSE, row.names = FALSE ) ######################### DEG ######################### group_info <- data.frame(Sample=samplelist, Group=c(rep("Col",3), rep("met1",3))) print(group_info) expr <- column_to_rownames(expr.mat, "GeneID") # 表达矩阵 head(expr) colData <- group_info # 分组信息 colData$Group <- factor(colData$Group, levels = c("Col", "met1")) dds <- DESeqDataSetFromMatrix(countData = as.matrix(expr), colData = colData, design = ~Group) dds <- dds[rowSums(counts(dds)) > 10, ] # 过滤低表达基因 dds <- DESeq(dds) # 差异分析 print("######## Compare groups") print(resultsNames(dds)) # 查看比较组别 print(dds) # log2(met1 / Col) res <- results(dds) # 获取差异表达结果 head(res) # 查看差异表达结果 ## 保存分析结果 write.csv(res, "./05.DEseq2/deseq2.diff.csv") ####################### Volcano ####################### res.plot <- data.frame(res, stringsAsFactors = FALSE, check.names = FALSE) res.plot <- na.omit(res.plot) res.plot$gene <- rownames(res.plot) res.plot$sig <- "Not Signaficant" res.plot[res.plot$log2FoldChange > 2 & res.plot$padj < 0.01, ]$sig <- "Highly expression in met1" res.plot[res.plot$log2FoldChange < -2 & res.plot$padj < 0.01, ]$sig <- "Highly expression in Col" res.plot$label <- ifelse(res.plot$padj < 10e-100 & abs(res.plot$log2FoldChange) >= 5, rownames(res.plot), "") p <- ggplot(data=res.plot, aes(x=log2FoldChange,y= -log10(padj))) + geom_point(aes(color=sig)) + geom_text(aes(label=label), nudge_x=.5,nudge_y=.5, size=2.5) + scale_color_manual(values=c("#546de5", "#ff4757", "#d2dae2")) + labs(x=expression(log[2](FC)), y=expression(-log[10](padj))) + geom_hline(yintercept=2, linetype=4) + geom_vline(xintercept=c(-2, 2),linetype=4) + theme_bw() + theme(panel.grid = element_blank(), legend.position = "top", legend.title = element_blank(), aspect.ratio=1, legend.background = element_blank()) ## 保存 ggsave("./05.DEseq2/Volcano.plot.pdf", p, width=5.5, height=5.5)
与原PPT有点差异,不知道原PPT是比对到全基因组,还是只是Chr1。
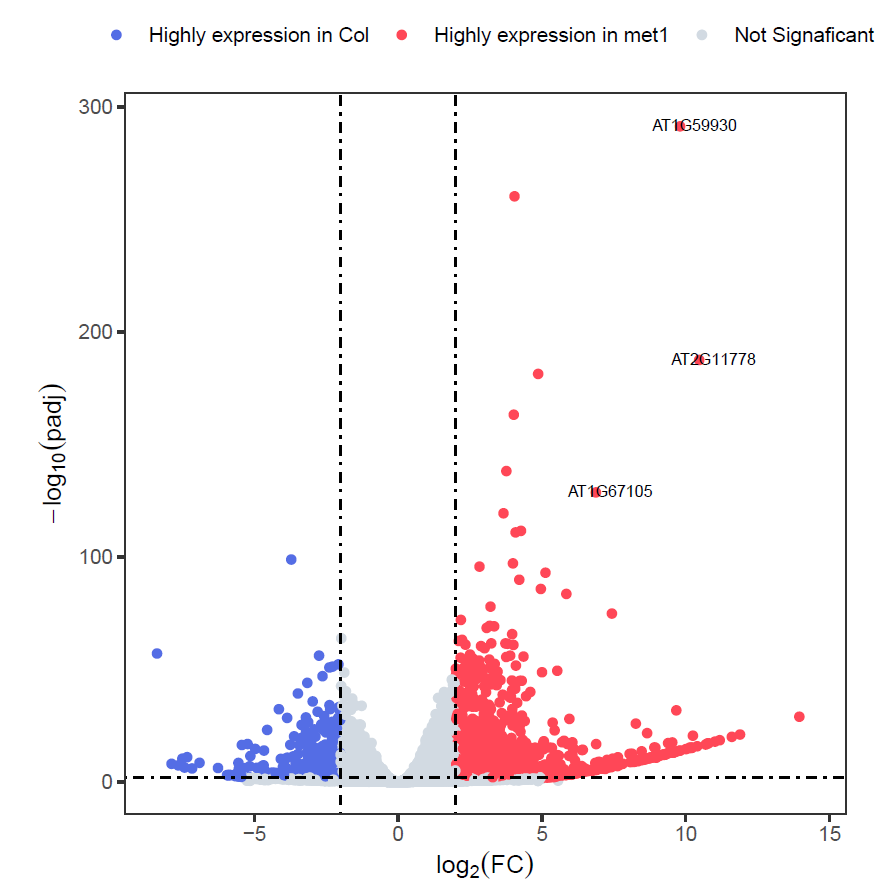
3. snakemake pipeline
Snakefile# Snakefile for RNA-seq analysis pipeline (with pre-provided reference) configfile: "config.yaml" # --- 从配置文件读取参数 --- WORK_DIR = config["work_dir"] THREADS = config["threads"] REF_FASTA = config["ref_fasta"] # 基因组fasta路径 REF_GTF = config["ref_gtf"] # 注释文件路径 REF_PREFIX = config["ref_prefix"] # 获取所有样本列表 ALL_SAMPLES = [sample for group in config["samples"].values() for sample in group] SAMPLES_GROUP = {sample:group for group in config["samples"] for sample in config["samples"][group]} GROUP_OF_SAMPLES = [SAMPLES_GROUP[sample] for sample in ALL_SAMPLES] GROUP_NAMES = unique_groups = list(dict.fromkeys(GROUP_OF_SAMPLES)) print(GROUP_NAMES) # --- 定义目录结构 --- dirs = [ "00.raw_sra", "01.raw_fastq", "02.clean_fastq", "03.alignment_file/hisat2_result", "04.count_matrix/featureCounts", "05.DEseq2", "Reference/hisat2_index" ] rule all: input: expand("05.DEseq2/{plot}", plot=["merged_count_matrix.txt", "deseq2.diff.csv", "Volcano.plot.pdf"]), "03.alignment_file/heatmap_SpearmanCorr_readCounts.png" # --- 创建目录结构 --- rule create_dirs: run: import os for d in dirs: os.makedirs(os.path.join(WORK_DIR, d), exist_ok=True) # --- 数据下载和质控部分保持不变 --- rule download_sra: output: "00.raw_sra/{sample}/{sample}.sra" params: outdir = "00.raw_sra" shell: "{config[tools][prefetch]} {wildcards.sample} -O {params.outdir}" rule sra_to_fastq: input: "00.raw_sra/{sample}/{sample}.sra" output: "01.raw_fastq/{sample}_1.fastq.gz", "01.raw_fastq/{sample}_2.fastq.gz" shell: "{config[tools][fastq-dump]} {input} --split-3 --gzip -O 01.raw_fastq" # --- FastQC原始数据质控 --- rule raw_fastqc: input: raw_fq1 = "01.raw_fastq/{sample}_1.fastq.gz", raw_fq2 = "01.raw_fastq/{sample}_2.fastq.gz" output: "01.raw_fastq/{sample}_1_fastqc.html", "01.raw_fastq/{sample}_2_fastqc.html", "01.raw_fastq/{sample}_1_fastqc.zip", "01.raw_fastq/{sample}_2_fastqc.zip" shell: "{config[tools][fastqc]} {input.raw_fq1} -o 01.raw_fastq -t 1;" "{config[tools][fastqc]} {input.raw_fq2} -o 01.raw_fastq -t 1" # --- MultiQC汇总报告 --- rule raw_multiqc: input: expand("01.raw_fastq/{sample}_{pair}_fastqc.html", sample=ALL_SAMPLES, pair=["1", "2"]) output: "01.raw_fastq/multiqc_report.html" shell: "{config[tools][multiqc]} 01.raw_fastq/ -o 01.raw_fastq" # --- Fastp数据清洗 --- rule fastp_clean: input: r1 = "01.raw_fastq/{sample}_1.fastq.gz", r2 = "01.raw_fastq/{sample}_2.fastq.gz", raw_multiqc = "01.raw_fastq/multiqc_report.html" output: cr1 = "02.clean_fastq/{sample}_1.fastq.gz", cr2 = "02.clean_fastq/{sample}_2.fastq.gz", html = "02.clean_fastq/{sample}.fastp.html", json = "02.clean_fastq/{sample}.fastp.json" shell: "{config[tools][fastp]} -i {input.r1} -I {input.r2} " "-o {output.cr1} -O {output.cr2} " "-h {output.html} -j {output.json} " "--thread 4" # --- 清洗后质控 --- rule clean_fastqc: input: clean_fq1 = "02.clean_fastq/{sample}_1.fastq.gz", clean_fq2 = "02.clean_fastq/{sample}_2.fastq.gz" output: "02.clean_fastq/{sample}_1_fastqc.html", "02.clean_fastq/{sample}_2_fastqc.html", "02.clean_fastq/{sample}_1_fastqc.zip", "02.clean_fastq/{sample}_2_fastqc.zip" shell: "{config[tools][fastqc]} {input.clean_fq1} -o 02.clean_fastq -t 1;" "{config[tools][fastqc]} {input.clean_fq2} -o 02.clean_fastq -t 1" rule clean_multiqc: input: expand("02.clean_fastq/{sample}_{pair}_fastqc.html", sample=ALL_SAMPLES, pair=["1", "2"]) output: "02.clean_fastq/multiqc_report.html" shell: "{config[tools][multiqc]} 02.clean_fastq/ -o 02.clean_fastq" # --- 参考基因组处理 --- print(REF_FASTA) rule hisat2_index: input: REF_FASTA output: expand("Reference/hisat2_index/{prefix}.{n}.ht2", prefix=config["ref_prefix"], n=range(1,9)) shell: "{config[tools][hisat2]}-build -p {THREADS} {config[ref_fasta]} Reference/hisat2_index/{config[ref_prefix]}" # --- 比对和定量 --- rule hisat2_align: input: clean_multiqc = "02.clean_fastq/multiqc_report.html", r1 = "02.clean_fastq/{sample}_1.fastq.gz", r2 = "02.clean_fastq/{sample}_2.fastq.gz", idx = expand("Reference/hisat2_index/{prefix}.{n}.ht2", prefix=config["ref_prefix"], n=range(1,9)) output: sam = "03.alignment_file/hisat2_result/{sample}.hisat2.sam", summary = "03.alignment_file/hisat2_result/{sample}.hisat2.summary" shell: "{config[tools][hisat2]} -x Reference/hisat2_index/{config[ref_prefix]} " "-1 {input.r1} -2 {input.r2} " "-S {output.sam} -p 4 " "--summary-file {output.summary}" rule sam_to_bam: input: sam = "03.alignment_file/hisat2_result/{sample}.hisat2.sam" output: bam = "03.alignment_file/hisat2_result/{sample}.hisat2.sort.bam", bai = "03.alignment_file/hisat2_result/{sample}.hisat2.sort.bam.bai" # 索引文件 params: unsorted_bam = "03.alignment_file/hisat2_result/{sample}.hisat2.bam" # 中间文件 shell: """ # 1. SAM转BAM {config[tools][samtools]} view -@ 4 -bS {input.sam} > {params.unsorted_bam} # 2. 排序BAM {config[tools][samtools]} sort -@ 4 {params.unsorted_bam} -o {output.bam} # 3. 删除中间文件 rm -v {params.unsorted_bam} {input.sam} # 4. 创建索引 {config[tools][samtools]} index {output.bam} """ rule feature_counts: input: bam = "03.alignment_file/hisat2_result/{sample}.hisat2.sort.bam", gtf = REF_GTF output: counts = "04.count_matrix/featureCounts/{sample}.featurecount.txt" shell: "{config[tools][featurecounts]} -a {input.gtf} -o {output.counts} " "-p {input.bam}" # --- 样本相关性分析 --- rule sample_correlation: input: bams = expand("03.alignment_file/hisat2_result/{sample}.hisat2.sort.bam", sample=ALL_SAMPLES) output: "03.alignment_file/heatmap_SpearmanCorr_readCounts.png", "03.alignment_file/readCounts.npz", "03.alignment_file/readCounts.tab" params: labels = lambda wildcards: " ".join( [f"{group}_rep{i+1}" for group in config["samples"] for i, sample in enumerate(config["samples"][group])] ) shell: "{config[tools][multiBamSummary]} bins --bamfiles {input.bams} " "--minMappingQuality 30 --labels {params.labels} " "-out {output[1]} --outRawCounts {output[2]} " "-p {THREADS} \n" "{config[tools][plotCorrelation]} -in {output[1]} --corMethod spearman " "--skipZeros --plotTitle 'Spearman Correlation of Read Counts' " "--whatToPlot heatmap --colorMap RdYlBu --plotNumbers " "-o {output[0]}" # --- DESeq2差异分析 --- rule merged_deseq2_analysis: input: counts = expand("04.count_matrix/featureCounts/{sample}.featurecount.txt", sample=ALL_SAMPLES), gtf = REF_GTF output: merged_count_txt = "05.DEseq2/merged_count_matrix.txt", diff_csv = "05.DEseq2/deseq2.diff.csv", volcano = "05.DEseq2/Volcano.plot.pdf" run: # 准备分组信息 group_data = [] for group_name, samples in config["samples"].items(): group_data.extend([f"'{s}':'{group_name}'" for s in samples]) # 将列表转换为字符串 sample_str = ','.join([f"'{s}'" for s in ALL_SAMPLES]) group_str = ','.join([f"'{g}'" for g in GROUP_OF_SAMPLES]) group_class_str = ','.join([f"'{c}'" for c in GROUP_NAMES]) # 生成R脚本 script = f""" library(purrr) suppressMessages(library(DESeq2)) library(ggplot2) library(tibble) samplelist <- c({sample_str}) grouplist <- c({group_str}) exprlist <- lapply(samplelist, function(x){{ expr <- read.table(paste0('04.count_matrix/featureCounts/', x, '.featurecount.txt'), header=T) expr<- expr[,c(1,7)] colnames(expr) <- c('GeneID', x) expr }}) expr.mat <- purrr::reduce(exprlist, function(x,y){{merge(x,y, by="GeneID", all.x=T)}}) write.table(expr.mat, file = "{output.merged_count_txt}", sep = "\t", quote = FALSE, row.names = FALSE) group_info <- data.frame(Sample=samplelist, Group=grouplist) print(group_info) expr <- column_to_rownames(expr.mat, "GeneID") head(expr) colData <- group_info colData$Group <- factor(colData$Group, levels = c({group_class_str})) dds <- DESeqDataSetFromMatrix(countData = as.matrix(expr), colData = colData, design = ~Group) dds <- dds[rowSums(counts(dds)) > 10, ] dds <- DESeq(dds) print(resultsNames(dds)) print(dds) res <- results(dds) head(res) write.csv(res, "{output.diff_csv}") res.plot <- data.frame(res, stringsAsFactors = FALSE, check.names = FALSE) res.plot <- na.omit(res.plot) res.plot$gene <- rownames(res.plot) res.plot$sig <- "Not Signaficant" res.plot[res.plot$log2FoldChange > 2 & res.plot$padj < 0.01, ]$sig <- "Highly expression in met1" res.plot[res.plot$log2FoldChange < -2 & res.plot$padj < 0.01, ]$sig <- "Highly expression in CK" res.plot$label <- ifelse(res.plot$padj < 10e-100 & abs(res.plot$log2FoldChange) >= 5, rownames(res.plot), "") p <- ggplot(data=res.plot, aes(x=log2FoldChange,y= -log10(padj))) + geom_point(aes(color=sig)) + geom_text(aes(label=label), nudge_x=.5,nudge_y=.5, size=2.5) + scale_color_manual(values=c("#546de5", "#ff4757", "#d2dae2")) + labs(x=expression(log[2](FC)), y=expression(-log[10](padj))) + geom_hline(yintercept=2, linetype=4) + geom_vline(xintercept=c(-2, 2),linetype=4) + theme_bw() + theme(panel.grid = element_blank(), legend.position = "top", legend.title = element_blank(), aspect.ratio=1, legend.background = element_blank()) ggsave("{output.volcano}", p, width=8, height=6) """ with open("merged_analysis.R", "w") as f: f.write(script) shell("Rscript merged_analysis.R")
config.yaml# config.yaml samples: CK: - SRR15859987 - SRR15859988 - SRR15859989 met1: - SRR15859993 - SRR15859994 - SRR15859995 # 参考文件配置 ref_fasta: "../Reference/Arabidopsis_thaliana.TAIR10.dna_sm.toplevel.fa" ref_gtf: "../Reference/Arabidopsis_thaliana.TAIR10.61.gtf" ref_prefix: "Arabidopsis_thaliana.TAIR10" work_dir: "." threads: 16 # 工具路径配置 tools: fastqc: "fastqc" multiqc: "multiqc" fastp: "fastp" hisat2: "hisat2" samtools: "samtools" featurecounts: "featureCounts" prefetch: "prefetch" fastq-dump: "fastq-dump" multiBamSummary: "multiBamSummary" plotCorrelation: "plotCorrelation"
snakemake -j 20